
原标题:糖尿病:过去、现在和未来(十):雄关漫道(下)
上一回我们讲到,至少从药物开发的角度,二型糖尿病是一种相比一型糖尿病更为复杂的疾病。后者由缺乏胰岛素导致,借助胰岛素注射能够得到有效的控制;前者由于身体对胰岛素失去响应导致,而如何重新唤醒我们的身体却是件非常复杂的任务。
恩斯特.史达林
英国科学家,激素的发现者和激素(hormone)一词的发明人。一个很有趣的小插曲是,史达林和贝里斯的实验,最初是试图证明俄国科学家巴甫洛夫的一个猜想,即胰腺消化液的分泌完全由神经所控制。不过史达林和贝里斯在实验中发现,切断神经并不能阻止胰腺消化液的分泌,证明他们最初的猜想是错误的。没有就此放弃的他们也因此转向分析究竟这背后是何种物质或刺激起作用,从而发现小肠产生了激素调节胰腺分泌。(图片来自Tata EMBO Reports 2005)
从这个时候看,我们的故事和糖尿病还没有一丁点的关系。
第一点联系来自于三十年后的1932年。此时人们已经相当清楚胰腺的两个彼此独立的功能:分泌消化酶和分泌胰岛素。既然小肠分泌的激素能够促进消化液的分泌,那么是不是也能促进胰岛素分泌呢?
受到这个想法的鼓舞,比利时科学家让?巴尔(Jean La Barre)重复了贝里斯和史达林的工作并发现,狗小肠分泌的激素确实具有降低血糖的功能。
不仅如此,巴尔成功地利用生物化学方法把小肠分泌物分成了两个组分,使它们分别能够促进消化液分泌和降低血糖,证明了至少有一种小肠激素分子能够专一的降低血糖。
但是在之后的几年,这类被巴尔命名为“肠泌素”(incretin)的物质却被同行发现效果很可疑:把肠泌素注射到糖尿病患者体内,根本看不到什么降低血糖的反应。肠泌素的概念,和它与糖尿病的可能关联,也因此被人迅速的遗忘,而且一忘就是又一个三十年。
在1960年代,随着技术的进步,人们得以能够直接检测和量化血液中含量极低的胰岛素分子,从而能够研究胰岛素水平的变化规律。例如,就像咱们故事里讲过的那样,喝一杯糖水之后人体血糖水平上升,同时伴随着胰岛素水平的上升。这时候人们发现了一个非常怪异的现象:如果同样一杯糖水不是被喝下,而是被直接注射到血液里,那么人体胰岛素水平上升的就要慢得多、少得多!
这个就太奇怪了。口服的葡萄糖要经过口腔、食管、胃,直到进入人的小肠,才能被吸收和进入血液循环,这个过程中的被动损耗暂且不提,单就时间而言,无论如何都应该比注射葡萄糖进入血管慢得多。
那么按照常理推断,注射葡萄糖刺激胰岛素分泌的能力应该要远远高于口服才对啊。
而“不合常理”的观察结果,往往是美妙发现的前奏。
就看当时那位屏住呼吸等待的观察者,是更愿意相信“自古以来”,“理当如此”,还是更相信理性的力量了。
亲爱的读者们,你们是哪种人呢?
我相信你们中的某些人,这时候已经想到了些什么:口服的葡萄糖能够更强有力地刺激胰岛素的分泌,这说明葡萄糖经过消化道的时候,会因为某种未知的原因刺激胰岛素分泌;反过来,绕开消化道直接进入血管的葡萄糖则没有这个本事。
且慢,这不恰好对上了巴尔医生1932年的观察和猜测么?
葡萄糖进入小肠-->小肠分泌“肠泌素”-->肠泌素刺激胰岛素分泌-->血糖降低?(在巴尔的发现的基础上,增加了“肠泌素刺激胰岛素分泌”这个新发现)
于是肠泌素的概念在三十年后被如获至宝地重新捡了回来。和巴尔医生的时代不同的是,此时的科学家已经有了更好的研究手段,其中之一就是我们之前讲过桑格蛋白质测序法。于是在二十世纪六七十年代,人们开始了从小肠分泌物中寻找传说中的肠泌素分子的竞赛。
很快,两种符合“肠泌素”定义的蛋白质分子被找了出来。
它们分别被命名为GIP(gastric insulinotropic peptide/葡萄糖依赖性粗胰岛素分泌多肽)和GLP-1(glucagon like peptide-1/胰高血糖素样多肽-1)。
读者们尽可以忽略这两个佶屈聱牙的名称,我们只需要知道,GIP和GLP-1两个蛋白质,都是从小肠肠壁细胞分泌并进入血液,都能够刺激胰岛贝塔细胞分泌胰岛素,就足够了。
这两种激素接近完美地解释了口服葡萄糖的古怪后果:葡萄糖进入小肠后能够刺激这两类激素的分泌,从而更好地刺激了胰岛素分泌和血糖的下降。
兴奋不已的科学家们第一个想到的就是:能不能用GIP和GLP-1治疗二型糖尿病(它们显然不能用来治疗一型糖尿病,因为一型糖尿病人根本没有贝塔细胞可以被刺激)?
毕竟两者和胰岛素一样,都是人体天然合成的蛋白质,安全性应该没问题。同时,对于二型糖尿病人而言,如果能够增强胰岛素的分泌,至少应该能够稍微缓解机体对胰岛素响应能力的下降,从而起到治疗的效果。
这时候巴尔式的失败又一次降临了:将GLP-1持续透析进入糖尿病患者体内的效果确实不错。但是注射GLP-1的效用虽然不能说完全没有,但是微乎其微,几乎没有什么临床意义。
不过这一次,有更好的蛋白质定量技术,人们很快地找到了原因所在(也正是为什么巴尔的实验长久以来无法被重复的原因):GIP/GLP-1在体内会迅速地被分解并通过肾脏排出,其半衰期只有惊人的一两分钟!
在这么短的时间内,再神奇的药也来不及唤醒胰岛素从而降低血糖啊。
肠泌素及其治疗糖尿病的希望,是不是就此退出历史舞台了?
没有,没有。
别忘了我们刚刚说的,“不合常理”的观察结果,往往是美妙发现的前奏。正因为这个令人沮丧的发现,肠泌素的故事从此进入快节奏,和命运多舛的二甲双胍分道扬镳。
“理性”制药和利拉鲁肽的诞生
肠泌素能被我们的身体降解这一发现,迅速为科学家们指明了一条能够摆脱“炼金术”,“理性”开发糖尿病药物的光明道路。
读者们不妨暂停阅读,给自己布置一点点思维体操的作业。如果你是药物开发者,该怎么利用这一个初看令人沮丧的发现呢?
一方面,肠泌素(特别是GLP-1)确实有很好的促进胰岛素分泌、降低血糖的效果。而另一方面,注射肠泌素仅有极短的生命期,难以起到治病救人的作用。
那么看起来,是不是至少有两个办法能解决问题?
一个可能性是有意识地修饰和改变GLP-1的结构,让它变得更“皮实”一点,不太容易被身体降解和排出;另一个可能性则是釜底抽薪,干脆找到身体里到底什么物质负责降解GLP-1,把它给抑制了不就行了?
从这两个思路出发,读者们也许能开始感受到所谓“理性”制药的含义。
在这里,我们不再需要依赖意外的观察和偶然的发现(比如山羊豆能够毒死牲畜)来提示我们某种潜在药物的存在,我们可以根据对生命现象的认知,主动地、有意识地去创造出我们需要的药物来。
先说说这前一个思路吧。目标非常明确:我们已经知道GLP-1这种肠泌素能够刺激胰腺分泌更多的胰岛素,我们需要的是尽可能的延长它在体内的半衰期,使其充分发挥功能。按照“炼金术”的思路,科学家们和药物开发者们大概需要在野外到处踅摸奇怪的现象,指望不定哪一天能从某种神秘动物的体内找到一种“恰好”可以在人体内活的久一点的GLP-1。
事实上人们确实也这么做了,第一种类似GLP-1的药物正是从一种从有毒的蜥蜴中发现的蛋白质,人们发现它在人体内的半衰期要比人类GLP-1长得多,于是就移花接木地拿它治疗二型糖尿病。
而利拉鲁肽,全世界第二个上市的类似GLP-1的药物,则更好地说明了“理性”制药的特点。和第一种类似GLP-1的药物不同,利拉鲁肽是人类原生GLP-1的衍生物;它也不是来自漫无目的地寻找,而是来自实验室中目的明确的设计。
它到底是怎么来的呢?
长话短说,利拉鲁肽的设计充分利用了科学界对GLP-1的各种研究成果。
人们知道,GLP-1是一个由三十个氨基酸组成的蛋白质,它之所以有着短得惊人的半衰期,是因为它在体内很容易被蛋白酶切割,并随即进入肾脏并被排泄。
因此,想要延长GLP-1的半衰期,关键是防止它被蛋白酶切割。
与此同时,人们通过对胰岛素的多年摸索,已经发现如果在蛋白质分子上连上一段长长的脂肪链,就有可能抵抗蛋白酶的进攻,延缓蛋白质被切割降解的速度。事实上一部分长效胰岛素就是根据这个思路制造出来的。
结合这两条,科学家们就可以大批量地尝试对天然GLP-1进行改造,特别是在30个氨基酸的基础上增加脂肪链,以期制造出能存活得更久的GLP-1类似物了。
可是拿到那么多类似物之后,怎么知道哪种真的有效呢?总不能每种都拿来往人身上注射了看效果吧?
而人们同时也知道,GLP-1之所以能够促进胰岛素分泌,是因为它能够特异地结合胰腺贝塔细胞上的受体蛋白。
因此,拿到一系列类似物候选分子之后,药物开发者们只需要在试管里检测这些分子和GLP-1受体的结合强度,就能够很好地预测哪种候选分子效用强劲了。
就是这样,2000年,诺和诺德公司的科学家们第一次报道了利拉鲁肽的合成和基本特性。在之后的十年中,利拉鲁肽经受了严苛的临床检验,并最终于2009和2010年在欧洲和美国上市(2011年在中国上市)。而就在今年,利拉鲁肽还在美国市场获得了作为肥胖症药物的资格。

利拉鲁肽的示意图
深红色是人类GLP-1蛋白原有的氨基酸(每一个红色圆圈是一个氨基酸,而GLP-1由三十个氨基酸相连组成)。利拉鲁肽相比人类GLP-1仅有两处差别:一个赖氨酸被替换成了性质类似的精氨酸(Arg-->Lys),另外在蛋白质中央附近连接上了一条由16个碳原子构成的脂肪酸链。读者们可想而知,对GLP-1的发现以及对其生物学的深入研究,是发明利拉鲁肽的基础。
笔者不是临床医生,也无意评价任何一个糖尿病药物的具体临床表现。笔者想展示给大家的,更多是一个摆脱了“炼金术”色彩的药物开发的故事。在这个故事里,药物开发者们在一开始就设定好了清楚的目标,通过理性的实验设计和临床验证,最终推出一种革命性的新药。
而读者们不应该忽略的是,许多代科学家们对人体奥秘的探索,一步步奠定了理性制药的基础。一百年来,来自实验室的发现,证明了激素的存在,提示和最终发现了神奇的肠泌素,揭示了GLP-1促进胰岛素分泌的原因,发现了GLP-1被迅速降解的秘密……
这些人类最聪明头脑的智慧结晶,最终使得利拉鲁肽的到来显得如此的水到渠成。
药物设计与锁匠的游戏
在试图改造GLP-1,让它变得更皮实和经久耐用的同时,人们还在尝试另一种“釜底抽薪”的制药思路。
既然GLP-1在体内半衰期极短,很容易被蛋白酶切割和降解,那么何不找出罪魁祸首是哪种蛋白酶,干脆将它破坏或者抑制掉?
这个思路说难不难,说简单却也没有那么简单。
说它不难,是因为早在1993年,人们已经知道了GLP-1是如何被降解的。
德国基尔大学的科学家们发现,GLP-1能在试管里被一种名叫二肽基肽酶-4(DPP-4/dipeptidyl peptidase-4)的蛋白酶切掉一端的两个个氨基酸,从而失去活性。这一发现也很快被动物体内的实验所证实。
因此从理论上来说,只要能找到一个办法,破坏掉DPP-4蛋白酶的活性,就能够延长GLP-1在体内的作用时间,从而达到治疗二型糖尿病的目的。
事实上,从DPP-4对GLP-1的切割功能发现的那一天开始,学术界,工业界的各路神仙就开始了针对DPP-4的攻坚战。
而说它不容易,是因为想要定点破坏掉身体中一个蛋白质的活性,并不是件一蹴而就的便宜事。这里面至少隐藏着两个需要克服的技术问题:第一,你怎么找到一个破坏其活性的办法?第二,你怎么能保证这个方法只破坏掉你感兴趣的蛋白质,而不会对身体里其它各种各样的重要蛋白质造成威胁?这两个问题一是关系到药物的药效,二是关系到药物的副作用,缺一则难成大器。
解决前一个药效问题有几个“理性”程度不等的思路。
比如说,一个办法是所谓的“高通量筛选”。
简单来说,就是把DPP-4蛋白酶放在试管里,然后把成千上万、甚至上百万的各种小分子化合物一个一个丢进去,看看哪一种能够有效的抑制其活性,找出来之后修饰修饰直接当药吃。
读者们还别笑,事实上尽管今天的药厂装备了各种自动化的高大上技术,但很多时候药物研发的第一步仍然是这种暴力美学的路子。
第二个开发思路叫做“构效关系”。
这个思路的逻辑是,利用手里已知的几个能够抑制DPP-4蛋白酶活性的小分子,然后比较这些小分子的化学结构,试图从中找到继续优化和改进小分子活性的办法。
打个比方,这个方法就像出门去买衣服,试了一件黄色的长裙觉得裙摆太长显得矮,又试了一件蓝色的短裙觉得蓝色太深不够活泼,那这位美女大概应该去找一件黄色的短裙能够满足她的挑剔眼光。
读者们可以看到,这种方法就有那么点“理性”的意思在里头了。
而今天笔者要展示给大家的,是相对来说最“理性”的一种办法,叫做“基于结构的药物设计”。
同时,这种方法还顺便把药物的特异性,也就是安全性的问题给解决了。倒不是说这种办法一定就比上面说的两种办法有效,而是这种办法最能体现现代药物设计所能达到的理性高度。实际上,DPP-4抑制剂类型的药物中最成功的例子,默克公司的捷诺维,反倒是上述两种暴力方法的产物。
具体来说,基于结构的药物设计的思路是这样的,对于任何一种酶来说,它能起作用的催化功能都是与其自身的三维立体结构相对应的。打个比方的话,一种酶和它的作用底物有点像钥匙和锁的关系。酶分子就像一把锁,只有特定性状的作用底物(也就是钥匙)才能插得进去并且转动锁,从而发挥功能。当然这个比方的结局带着点黑色幽默的色彩:对于蛋白酶和底物来说,钥匙转动锁(底物插进蛋白酶)的后果就是钥匙“咔嚓”一声被掰断了(底物被蛋白酶切割)。
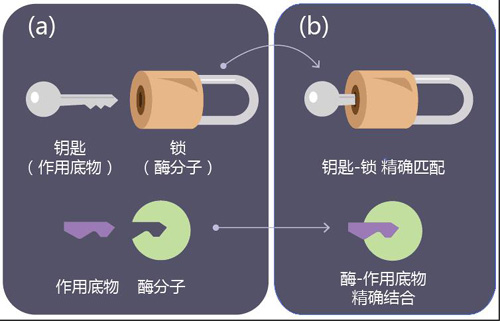
解释酶分子功能的“锁与钥匙”模型(绘图 肖媛)
我们知道,锁和钥匙需要配对才能开锁。相似的,一个酶分子(绿色)和锁一样,也具备某种的构象,只能特异地识别某种底物分子(紫色),两者精确结合才能激活酶的功能。在DPP-4的故事里,DPP-4就像一把锁一样特异地识别GLP-1分子的形态,并切割GLP-1一端的氨基酸。不过要提醒读者们注意的是,锁与钥匙模型仅仅是对酶分子功能的粗浅解释,酶的反应动力学要复杂得多。比如,和锁不同的是,酶分子的构象会根据底物的不同而发生可塑的变化。
因此,按照这个理解,药物开发就有点像锁匠的游戏。如果我们能仔细描画出DPP-4蛋白酶这把锁的细微结构,就能够制造出一把坚固无比不会被掰断的钥匙来。
这样一来,这把钥匙就能够牢牢地占据锁孔不再离开;其他的钥匙,包括GLP-1,也就找不到机会开锁,或者说被掰断了。这样的话,DPP-4蛋白酶的活性就被抑制,而GLP-1的生命周期也就延长了。
2003-2004年,数篇学术论文集中报道了DPP-4蛋白酶的三维晶体结构(值得注意的是其中大部分研究来自于开发糖尿病药物的公司,例如诺和诺德、罗氏等等),从而让人们第一次清楚的看到了DPP-4这把锁的细节。
人们发现,DPP-4蛋白相对光滑的表面有一个小小的口袋状的凹陷(叫做S1口袋/S1 pocket),这个口袋底部带有强烈的疏水性,因此可以恰到好处的把GLP-1末端的两个疏水氨基酸给“装”进去,然后再“咔嚓”一声切掉。

DPP-4蛋白酶的三维晶体结构
在这张图中蛋白质表面的带电情况被标注出来(红色-负电,蓝色-正电)。可以清楚看到,在总体比较光滑的蛋白质表面,有一个深红色的凹陷,这里就是我们讲到的S1口袋,也正是DPP-4蛋白酶结合GLP-1的位点所在。如果能够设计出一个小分子恰好填在S1口袋之中,也许就可以阻止DPP-4对GLP-1的切割。(图片来自Rasmussen et al Nature Struc Biol 2002)
有了锁的图案,药物开发者们就开始玩锁匠的游戏了:对照S1口袋的大小、深浅和形状,把不同的小分子的图片往里面搁,看哪个更适合当钥匙。如果现成的小分子不够用,化学家们还可以在上面缝缝补补,左边加一个基团填补漏洞,右边换一个原子增加吸力之类。要知道,这一切工作可以在电脑上虚拟完成,因此可以以迅雷不及掩耳盗铃的速度尝试几十万上百万的小分子图片。也正是用这个思路,美国Syrrx公司的科学家们设计出了一种结构上全新的糖尿病药物(这家公司之后被日本武田制药收购),并于2010年于日本上市。

基于结构的药物设计
美国Syrrx公司的科学家根据DPP-4蛋白酶S1口袋的性状和物理化学性质,在计算机上筛选了大量小分子,并从中找到一个能够很好的填充S1口袋,阻止DPP-4结合GLP-1的小分子。最终基于这个小分子,他们开发出了阿格列汀。在上面的图片里模拟了一个小分子填充进凹陷的S1口袋。(图片来自Feng et al J Med Chem 2007)
雄关漫道
由此读者们可以看到,糖尿病药物的市场折射出现代药物开发的多张面孔。就在你们阅读这篇文章的时候,全世界的糖尿病患者正在使用各种各样的药物改善自身的健康。
胰岛素的发现受到一百五十年前胰腺切除导致糖尿病的偶然观察所启发,并最终于上个世纪二十年代被发现和应用于临床;而其后蛋白质测序以及重组DNA技术的兴起,又把胰岛素的临床应用推进到新的高度,人们开始有意识的通过基因工程改造胰岛素,以期实现对血糖灵活和长期的控制。
而在一型和二型糖尿病的区分被明确之后,人们在胰岛素的辉煌中没有忘记持续寻找专注于二型糖尿病治疗的药物。二甲双胍的发现来自于对有毒牧草的偶然研究,从发现到临床走过了半个多世纪的漫长岁月。而其它种类的糖尿病药物,特别是我们刚刚讲到的利拉鲁肽和阿格列汀,它们的发现建立在人们对肠泌素生理功能的长期研究的基础上,从而显得更具理性、目的性,也能够更快地推进到临床应用之中。
而这显然不是一切的结束。
尽管有着上百年不懈的研究,有着种类繁多的药物选择,我们还不得不承认,糖尿病仍然是一种可以控制和管理,但却无法治愈的慢性顽疾。尽管有药物的帮助,糖尿病人的生活仍然需要接受严格的控制,而慢性糖尿病引发的各种并发症(例如我们讲到过的糖尿病肾病和糖尿病眼底疾病)至今仍然是我们难以攻克的堡垒。
雄关漫道真如铁,而今迈步从头越。
然而我们有理由乐观,因为也就是在此时此刻,有许许多多人类的英雄们在努力工作。他们的目标,也许是一种治疗疾病的灵丹妙药,也许是对一种疾病的更深理解,也有可能是一种纯粹对未知世界的好奇心……这些不同方向的努力涓滴汇流,一定会带给我们更健康的自己,更美妙的生活。
敬请期待下文《糖尿病:过去、现在和未来(完结):新地平线》。在整个故事的结尾,笔者愿意放下历史,和读者们一起展望一下我们可能拥有的美好未来。

“科普中国”是中国科协携同社会各方利用信息化手段开展科学传播的科学权威品牌。
本文由科普中国融合创作出品,转载请注明出处。
